Revista de la Facultad de Ciencias
Agrarias. Universidad Nacional de Cuyo. Tomo 55(1). ISSN (en línea) 1853-8665.
Año 2023.
Original article
Serological relationships among strains of grapevine
leafroll-associated virus 4 reflect the evolutive behavior of its coat protein
gene
Las
relaciones serológicas y la identidad molecular de variantes de grapevine
leafroll-associated virus 4 reflejan el comportamiento evolutivo del gen de su
proteína de cubierta
Melisa Lanza Volpe 1
Nancy Setien 1
Olga Gracia 1
Oscar Grau 2
1 EEA Mendoza INTA, San
Martín 3853. Luján de Cuyo (5507). Mendoza. Argentina.
2 IBBM
CONICET. UNLP. 49 y 115 s/n. La Plata (1900). Buenos Aires. Argentina.
Abstract
This research studied
serological relationships and genetic diversity of Argentinean isolates of
grapevine leafroll-associated virus-4 (GLRaV-4). Phylogenetic analysis of coat
protein (CP) sequences from 19 local isolates revealed clustering with the
previously described GLRaV-4 strain 5, strain 6, and strain 9 groups.
Evolutionary sequence analysis of the obtained and database-available sequences
showed evidence of recombination events. Additionally, both CP N- and
C-terminal regions appeared to be under purifying selection, but the N-terminal
region presented seven sites under positive selection, with a dN/dS
ratio 5-fold greater than that of the C-terminal region.
Serological reactivity against monoclonal antibodies supports a higher
occurrence probability for linear epitopes in the N-terminal region, as
inferred by the sequence analysis. The obtained results reflect an unusual
evolutionary behavior of the CP that, together with protein serological
reactivity, suggests biological significance of the observed variability.
Keywords: Molecular characterization and
serology; Grapevine leafroll disease; Ampelovirus; Selection
pressure; Antigenic properties.
Resumen
Las relaciones serológicas
y la diversidad genética de cepas argentinas de grapevine leafroll associated
virus 4 (GLRaV-4) fueron analizadas. El análisis filogenético de la cápside
proteica (CP) conducido sobre las secuencias obtenidas mostró un agrupamiento
de las primeras con GLRaV-4 raza 5, GLRaV-4 raza 6 y GLRaV-4 raza 9. El
análisis evolutivo de las secuencias locales y las disponibles en bases de
datos infirió eventos de recombinación y sugirió que tanto los extremos
C-terminal como N-terminal de la CP están bajo presión de selección
purificante, pero la región N-terminal mostró siete sitios bajo presión de
selección positiva, con una relación dN/dS cinco veces mayor que
aquellas posiciones de la región C-terminal. La reactividad serológica contra
anticuerpos monoclonales sustenta la probabilidad de ocurrencia de epitopes
lineales en la región N-terminal inferida en el análisis evolutivo. Los
resultados obtenidos reflejan un comportamiento evolutivo inusual de la CP y
junto con la reactividad serológica de dicha proteína, permiten postular una
significancia biológica de dicha variabilidad.
Palabras clave: Caracterización molecular y serología; Enfermedad del enrollado de la hoja de la vid; Ampelovirus; Presión
de selección; Propiedades antigénicas.
Originales: Recepción:
28/09/2022
Aceptación: 08/05/2023
Introduction
Grapevine Leafroll Disease
(GLD) is one of the most widespread and deleterious viral diseases affecting
grapevines with a particularly complex etiology. The development of serological
reagents led to the identification of seven putative species belonging to the
Closteroviridae family and generically named grapevine leafroll-associated
virus (GLRaV) 1-7. However, during the 1990s and 2000s, the easier acquisition
of genetic data arose the number of new putative species to 12 GLRaVs,
tentatively or definitively assigned to the Ampelovirus genus (most of
the species), to the Closterovirus genus (GLRaV-2) or the newly defined Velarivirus
genus (GLRaV-7) in the Closteroviridae family. Generally, the main
criteria establishing newly described viral isolates as new species were,
first, the lack of serological reaction against previously developed monoclonal
antibodies or polyclonal antiserum, and second, over 10% divergence in the
sequence of taxonomic relevant genes (HSP70h and CP). The proliferation of
taxonomic entities required a revision of the GLRaVs taxonomy, and considering
available genetic information, it was established that most of the GLRaVs
described at an early stage (GLRaV-4, -5, -6, -9, -De, -Pr and -Carn) should be
considered as divergent isolates of a single species (GLRaV-4) (21).
In addition to the
above-mentioned taxonomic issues, the serological relationships among the
GLRaV-4 groups of isolates (previously known as different species) remain
unclear. Gugerli (2009) performed an extensive
review of the different serological reagents developed during the past 30 years
and highlighted some of the arisen ambiguities concerning antibodies and
antisera. However, scarce information followed this review, being a serological
characterization of four isolates of GLRaV-4 (1) the most comprehensive work up to date. Western blot reactivity
among those four isolates (two of GLRaV-4 strain 6 and one of each strain 4 and
5) against monoclonal antibodies (Mabs) raised against GLRaV-4 strain 4, 5 and
6, was clear and straight, without any cross-reaction among them. Meanwhile,
all four isolates reacted against a monoclonal mix developed for generic
detection of GLRaV-4 (2) and a commercial
polyclonal antiserum anti-GLRaV-4 strain 5.
The complete genome
sequencing of different GLRaV-4 strains allowed clarifying the taxonomy of this
virus. Nevertheless, the significance of CP antigenic properties of the
different GLRaV-4 isolates remains unclear.
The present work presents a sequence analysis of the CP gene of
Argentinean GLRaV-4 isolates, and an additional serological analysis of
purified virions, aiming to establish significance levels of such genetic and
serological variability.
Material and Methods
Virus Isolates
One hundred forty-one
grapevine plants of different varieties exhibiting mild to severe symptoms of
grapevine leafroll disease (GLD) were selected from the ampelographic
collection located in the Mendoza Research Station of the National Institute of
Agronomic Technology (EEA Mendoza, INTA). In order to
determine symptomatic nature, these plants were analyzed by Enzyme-Linked
Immunosorbent Assay (ELISA) using commercial reagents for GLRaV-1, -2, -3, -4,
-4 strain 6, and -7. The study included positive samples for GLRaV-4 or GLRaV-4
strain 6 (24 samples) and those negative for all tested viruses (13 samples).
RNA extraction, RT-PCR, cloning, and sequencing
Double-stranded RNA
(dsRNA) was extracted from cambial scrapings of mature grapevine canes,
reverse-transcribed, and subsequently amplified by Polymerase Chain Reaction
(PCR) using a proofreading polymerase (DeepVent DNA Polymerase, New England
Biolabs, USA). The complete Coat Protein (CP) Open Reading Frame (ORF) of
GLRaV-4 was amplified using specific primers (ACPF
5’-GCTGGATAGGTTYAGRTCNAAAGAYACYCC-3’ and ACPR 5’-TAACCTCCATATTTTCAAACG-3’)
designed over the upstream and downstream sequences of such ORF (p60 and p23,
respectively) based on database available nucleotide sequences of GLRaV-4. The
resulting PCR products were resolved by agarose gel electrophoresis. The
occurrence of multiple infections with GLRaV-4 genetic variants in a single
plant was investigated by RT-PCR-Restriction Fragment Length Polymorphism
(RFLP), digesting the resulting PCR products with both AluI and HinfI
restriction enzymes. Restriction fragments were resolved by electrophoresis on
a 2% agarose gel. Undigested PCR products were cloned into the pGEM-T Easy
Vector System I, and the resulting clones were sequenced. After blue/white screening,
19 white colonies from each transformation were selected, amplified with ACPF
and ACPR, and restricted as mentioned for accurate identification of clones of
different genetic variants. Three colonies belonging to each restriction
pattern by sample were randomly selected and sequenced using both pUC/M13
reverse and forward sequencing primers at Macrogen Inc. (Korea).
Sequence analysis
Sequences from each clone were
assembled and edited obtaining the coding sequence of the CP ORF. Codon multiple
sequence alignment was performed using the aligned codons from these sequences,
together with all the GLRaV-4 CP sequences available in the NCBI GenBank
database. Using the HyPhy software package, evolutionary and phylogenetic
analyses evaluated whether selection pressure affects viral strain evolution (16). Recombination analysis was
performed using Single Breakpoint (SBP), Genetic Algorithm Recombination
Detection (GARD), and confirmed by RDP software (19). Three methods, namely Single
Likelihood Ancestor Counting (SLAC), Random Effects Likelihood (REL), and Fixed
Effects Likelihood (FEL), allowed the identification of selection pressure.
Results were integrated by integrative selection analysis. The aligned codons
(with additional CP sequence of PMWaV-1 (AF414119) as outgroup) were subjected
to preliminary phylogenetic reconstruction by Maximum Likelihood (ML) analysis
using the PAUP software package (28) and heuristic search, with
random addition sequences considering one hundred replicates. Group support was
estimated by Garli program (31) generating 1000 replicates to
obtain bootstrap values. Branches with bootstrap values under 70% were
collapsed. From the phylogenetic tree inferred, seven clusters were defined.
Sequences belonging to the different strains of GLRaV-4 allowed a clear
identification of such groups: GLRaV-4 strain 5 (AF233934), strain 9 (AY297819),
strain 6 (FJ467504), strain 4 (FJ467503), strain Ob (AB720874), strain Carn
(FJ907331), and strain Pr (FM244690). The genetic distance within and among
groups was estimated using the Tamura-Nei model of MEGA5 software by estimating
the standard error from a bootstrap of 1000 replicates. Overall distance of
GLRaV-4, and genetic distances for all the available CP sequences of GLRaV-1,
-2 and -3 were also estimated.
The presence of putative
linear epitopes over the deduced amino acid CP sequence was evaluated by
BepiPred software (18). Complementary to epitope
detection and through the SomeNA and SNAP2 tools implemented in the Predict
Protein server, a structure prediction analysis identified a putative nucleic
acid binding motif and the functional effect of a point mutation in CP (30).
Serological characterization
After the ELISA-positive
samples for GLRaV-4 or GLRaV-4 strain 6, nineteen samples were selected for
further serological assays based on the restriction pattern of observed CP.
Viral particles were purified from cortical scrapings of mature grapevine canes
as described by Savino (1993). The resulting
extracts were analyzed to determine serological characteristics of the GLRaV-4
variants. The purified virions were resolved over 30 mm wide lanes into a
14%/4% SDS PAGE, electroblotted to nitrocellulose membrane. After blocking the
membrane, ten individual longitudinal strips from each lane were excised. Each
strip (3 mm wide) was probed with each of seven monoclonal antibodies: Mab
36-117, Mab3-1, Mab8-2, Mab43-1, Mab3-3, Mab6-3, Mab 15-5 (12, 13) and three polyclonal antisera: ASGLRaV-5
from Biorad (Hercules, CA, USA), AS GLRaV-4 strain-6 from Bioreba AG
(Switzerland) and AS GLRaV-4 I252-IL (provided by Dr.
Boscia, 2006). The strips were revealed after incubation with
Goat-AntiMouse AP conjugated or Goat-AntiRabbit AP conjugated (Sigma, MI, USA).
Results
Out of the 141
ELISA-tested samples exhibiting leafroll disease symptoms, 13 samples reacted
with none of the tested reagents. When considering reactions against GLRaV-4
and GLRaV-4 strain 6 reagents, 10 samples resulted positive with both
antibodies, 12 only reacted with the GLRaV-4 reagents, and 2 samples only
reacted with the GLRaV-4 strain 6 reagents. These 37 samples were examined by
RT-PCR using the primers described above. No product was amplified from the 13
ELISA-negative samples, while a product of the expected size (1,100 bp) was
generated from all 24 ELISA-positive samples.
Restriction of the RT-PCR
products yielded several fragments in all cases Supplementary
Figure 1). In some cases, band number and size indicated presence of a
single genetic variant, while in other cases, the digestion of multiple PCR
products had the same size but different sequence. Cloning and screening of
these RT-PCR products using PCR and restriction over the white colonies allowed
to identify the genetic variants in the original sample. When two samples
shared the same restriction pattern and serological behavior, only one was
sequenced. Three colonies corresponding to each restriction pattern were
sequenced from samples with multiple patterns, considering a total of 90 clones
sequenced from 19 different plant samples. Generally, sequences of clones
sharing the same restriction pattern obtained from the same plant were
identical and considered a single sequence. Consequently, this study generated
30 sequences Supplementary Table 1). One single ORF was
identified in each sequence, sizing according to GLRaV-4 CP. Most sequences
produced a 269-amino acid translation product, but some sequences exhibited
minor size divergences (individual sequences of 265, 268, 271, and 272 amino
acids). Codon multiple alignment revealed deletions in the first 40 amino acids
of the protein (shorter sequences) or a mutation in the stop codon leading to
size differences.
Sequence analysis
The 164 sequences of GLRaV-4 used
considered 30 CP sequences obtained in this study and 134 sequences available
in NCBI Genbank database. The phylogenetic tree inferred by ML analysis
discriminated seven monophyletic groups supported by high bootstrap values Supplementary Figure 2). The seven GLRaV-4 described strains
(strain 4, strain 5, strain 6, strain 9, strain Ob, strain Pr, strain Carn)
represent the seven groups. This phylogeny agrees with the previously reported
phylogeny of the HSP70 of GLRaV-4 (20). Most of the local sequences
obtained in this study clustered with the reference sequences of GLRaV-4 strain
5 and GLRaV-4 strain 6, while only one sequence grouped with GLRaV-4 strain 9.
No local sequence clustered with GLRaV-4 strain 4, strain Pr, strain Ob or
strain Carn.
Table 1 shows the amino acid sequence identity level within and
among these seven groups.
Table 1: Estimates of evolutionary divergence between coat protein
sequences.
Tabla 1: Estimaciones de divergencia evolutiva entre secuencias de
proteínas de cápside proteica.
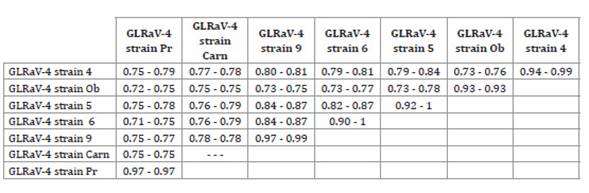
Range of aminoacidic identities per site between GLRaV4 sequences
of the seven groups identified and defined according to the inferred phylogeny.
Rango de
identidades aminocídicas por sitio entre secuencias de GLRaV-4 de los siete
grupos identificados y definidos según la filogenia inferida.
Identity among sequences
inside all these groups, except GLRaV-4 strain 6, exceeded 90%. Sequences
belonging to GLRaV-4 strain Ob and GLRaV-4 strain Pr were the most divergent,
sharing identities under the proposed 25% divergence threshold (21), with sequences from other groups. However, when the alignment
was arbitrarily split into two (first 40 residues, and from residue 40 to the
end of the protein), identity levels among sequences changed substantially. The
C-terminal region was conserved among all analyzed sequences. Identity level in
this region within the seven groups was over 95%, while identity level among
sequences of the different groups was always over 80% (Table 2).
Table 2: Estimates of evolutionary divergence between coat protein sequences
considering amino-terminal and carboxyl-terminal regions.
Tabla 2: Estimaciones de divergencia evolutiva entre las secuencias de
proteínas de cápside considerando las regiones amino-terminal y
carboxi-terminal.
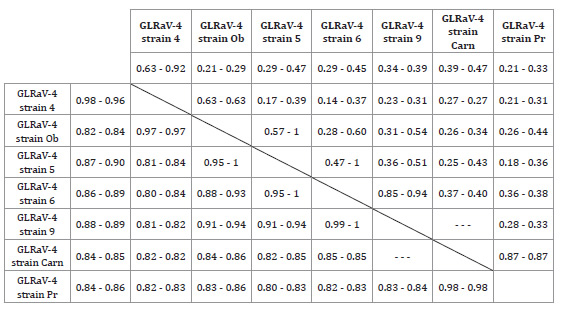
Range of aminoacidic identities per site between GLRaV4 sequences
of the seven groups identified and defined according to the phylogeny inferred.
Above the diagonal, comparison of the first 40 aminoacids. Below the diagonal,
comparison from residue 41 to the end of the sequences.
Rango de
identidades aminocídicas por sitio entre secuencias de GLRaV-4 de los siete
grupos identificados y definidos en base a la filogenia inferida. Por encima de
la diagonal, la comparación de los primeros 40 aminoácidos. Por debajo de la
diagonal, comparación desde el residuo 41 hasta el final de la secuencia
proteica.
Variability was
considerably high in the N-terminal region of CP with identity difference
levels between pairs of sequences as high as 43% within the GLRaV-4 strain 5
group, and 53% within the GLRaV-4 strain 6 group (Table 2). Average genetic distances estimated for the 128 CP
sequences of GLRaV-4 was 0.127, higher than GLRaV-3 but in line with GLRaV-2
(0.118) and the estimated 0.106 for GLRaV-1 (7). Both GLRaV-1 and GLRaV-2 are considered highly variable
viral species.
Evolutionary analysis
The recombination analysis
conducted by SBP inferred a putative recombination event over the multiple
sequence alignment. Such event was also detected by using the RDP software,
suggesting that GLRaV-4 strain 9 sequences were recombinants between GLRaV-4
strain 5 and GLRaV-4 strain 6. This same event was also identified when the
recombination analysis was performed with complete genomic sequences of GLRaV-4
strain 5, 6 and 9 available in the database. However, GARD was unable to detect
the recombination event among these three complete sequences. In consequence
and to avoid a biased analysis due to the recombination effect in the selection
pressure analysis, the complete alignment was split into two datasets according
to the inferred breakpoint (from position 1 to 120, and from 121 to the end of
the codon alignment).
Consequently, selection
pressure analysis was performed for two datasets. Results of overall dN/dS ratios obtained by SLAC and
REL were consistently different for both datasets (Table 3), being the mean dN/dS
ratio estimated by SLAC 5 times greater in the N-terminal region.
Table 3: Estimates of selection pressure on the coat protein gene of
GLRaV-4.
Tabla 3: Estimación de la presión de selección actuante en el gen de la
cápside proteica de GLRaV-4.
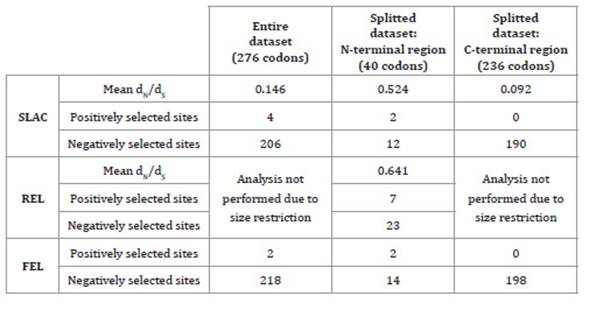
Although all obtained
ratios were lower than 1, indicating a negative or purifying selection, the
C-terminal region (the most conserved region) was subjected to heavier
purifying selection than the N-terminal region. When the dN/dS
ratio was analyzed for the entire CP as a single dataset, the
value fell in between the partial values obtained, similar to those reported by
Maliogka et al. (2008) (mean of 0.085 by
FEL). The site-by-site analysis integrating the three individual analyses (REL,
FEL, and SLAC) was different between both datasets. In the first dataset, seven
sites were significantly inferred as being under positive selection by REL (two
of them also identified by FEL and SLAC, Supplementary Figure
3), and 23 of the 33 remaining sites resulted under negative selection
pressure by at least one method. For the second dataset (position 121-end),
both methods failed to detect positive selection pressure, whereas 198 sites
(over 236 codons) were inferred as being under negative selection by FEL and
190 codons by SLAC. REL could not be performed due to alignment size
restrictions. In general, except for the negatively selected sites at positions
1, 5, 12, 14 20 and 21, the high prevalence of negatively selected sites begins
at position 24 of the multiple codon alignment (Supplementary
figure 3).
The linear epitope
prediction analysis of all concerned sequences performed by BepiPred revealed
the highest probability of occurrence of a B-cell linear epitope in the first
40 amino acids, in agreement with previous reports (6,
20). Noteworthy is that in the same positions, most of the sites
under positive selection were inferred, and as previously observed, the region
was the most CP variable (Supplementary Figure 3). In
every protein sequence, a single polynucleotide binding site was inferred by
SomeNA. All these predicted sites were located between the 175 to 182 CP
residues (Supplementary Figure 3).
Serological analysis
Western blot analysis of purified
extracts from nineteen GLRaV-4 infected plants and a virus-free accession
revealed variable specificity from the different Mabs and AS used. Two of the
three AS used (AS-GLRaV-5 and AS-I-252-IL) showed nonspecific reactions.
Several bands were observed in all the analyzed samples, even in the virus-free
Chardonnay. However, the 35KDa GLRaV-4 CP band was clearly identified. The
three AS reacted with the GLRaV-4 CP of the nineteen analyzed extracts, but the
Sangiovesse Fiano sample only faintly reacted with AS-I252-IL. Two Mabs (6-3
and 15-5) did not react with any western blot sample. The five remaining Mabs
showed variable reactions with the tested samples, from clear to faint bands (Figure
1).
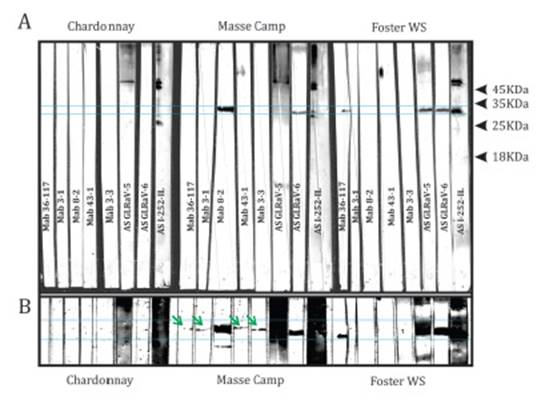
A) Reaction of three samples against five Mabs and three PAS.
Healthy controls clearly show nonspecific reactions against two PAS. B)
Overexposure of the same membrane reveals faint reaction against four Mabs.
A)
Reacción de tres muestras contra cinco anticuerpos monoclonales y tres
antisueros policlonales. El control sano muestra reacción no específica contra
dos antisueros. B) Sobre exposición de la misma membrana muestra reacción débil
contra cuatro anticuerpos.
Figure 1: Nitrocellulose membrane after western blot analysis.
Figura 1: Membrana de nitrocelulosa revelada tras western blot.
Western blot results are
summarized in Supplementary Table 1.
Discussion
Despite the taxonomic controversy
during the early ampelovirus history, today GLRaV-4 is considered a single
viral species composed of several genetic variants. In the present study, 30 CP
sequences of GLRaV-4 were obtained from 19 leafroll-affected grapevine plants.
All plants reacted with one or both ELISA reagents for GLRaV-4 and GLRaV-4
strain 6. The RT-PCR analysis of dsRNA extract from these plants allowed
amplifying a 1.100 bp fragment in all cases, containing the entire CP ORF.
Sequence analysis led to sequence identification of GLRaV-4 strain 5, strain 6
and strain 9. Serological analysis showed specific reactions of such samples
against Mabs for GLRaV-4 strain 5 (Mab 8-2, 43-1, 3-3) and -6 (Mab 36-117),
whereas no clear reaction was obtained against GLRaV-4-specific Mab (Mab 3-1).
Mab 15-5 and 6-3 did not react against any sample in western blot (as
previously recorded with the same extract used for mice immunization) (2). As these two Mabs compose the
GLRaV-4 DAS ELISA reagent set used, and give positive reaction with the tested
samples, both Mabs are obviously directed against a conformational epitope,
dissociated during the denaturing SDS-PAGE. Both molecular and serological analysis
revealed the occurence of mixed infections, an usual behavior in
grapevine-infecting closterovirids, also reported in other host species (8).
CP sequence variability of
GLRaV-4 was considerably high. In fact, some CP sequences (such as GLRaV-4
strains Ob and Pr) showed higher divergence than the proposed threshold of 25%
as criteria for species discrimination in the Ampelovirus genus (21). Given these variability levels, the proposed divergence
threshold of 25% over the CP aminoacidic sequence should be closely revised and
raised to 30% considering the currently available sequences belonging to
GLRaV-4. A closer analysis of such variability showed an asymmetrical
distribution, with the CP N-terminal region (the first 40 residues) much more
variable than the C-terminal region. Besides the nucleotide substitutions
leading to amino-acid changes, all insertions or deletions were concentrated in
the first 120 nucleotides of the multiple sequence alignment. Furthermore, a
segmented analysis of the observed variability evidenced that all available CP
sequences of GLRaV-4 shared more than 80% identity over the C-terminal region
(233 to 236 residues). In all cases, pair-wise similarity among sequences of
the referred C-terminal region was over 90%, reflecting a high conservation
degree.
The ML analysis performed
on the complete CP sequences herein generated and on the ones available in the
database revealed strong clustering in seven genetic groups. Most viral strains
obtained from local vineyards clustered together with GLRaV-4 strains 5 and 6,
while one of the obtained sequences exhibited a close relationship with GLRaV-4
strain 9. The seven groups of sequences were compared by a genetic distance
study conducted by using observed cluster distribution in the ML tree.
Intragroup level of genetic distance (≤ 0.1) was different from intergroup
distance (over 0.14 for the closest GLRaV-4 strain 5 and 9, and exceeding 0.2
for the remaining groups). These results are consistent with the amino acid
sequence identity levels between groups (Table 1). When GLRaV-4 was considered
as a single species, the average genetic distance in the group was 0.127,
barely higher than that in GLRaV-1 and -2, members of Ampelovirus and Closterovirus
respectively, and reported as highly variable species.
In the first evolutionary
analysis, a putative recombination event was inferred as related to GLRaV-4
strains 9, 5 and 6, producing a topological incongruence between the N- and
C-terminal regions of CP in the phylogenetic analysis. This difference was
already observed in the HSP70h and CP inferred phylogeny conducted by Maliogka et al. (2008), even though the authors
did not record any recombination evidence. Recombination is one main force
driving evolutionary history of plant viruses, with a significant impact in the
Closteroviridae family, as observed for CTV (Closterovirus),
GLRaV-3 and GLRaV-4 strain 5 (Ampelovirus). Considering GLRaV-4, the
restricted host range, long host lifespan, high sequence similarity and high
occurrence of mixed infections could provide a favorable environment for
increasing effective recombinants. However, the recombination signal identified
in GLRaV-4 could not constitute a true genetic exchange among donor and
recipient viruses, but a variable rate of mutation among the different genes of
the virus (29). A more detailed study
with more GLRaV-4 strain 9 sequences could confirm this event.
After splitting the dataset
according to putative recombination, a significant difference was revealed in
the selection pressure over CP during GLRaV-4 evolution history. In fact, the
entire CP sequence was subjected to a strong negative selection as indicated by
the global dN/dS
value of 0.144 (in concordance with most plant viral CP), but the
variable N-terminal region appeared to present sites subjected to positive
selection (fairly unusual in CP of plant virus). So far, the occurrence of
positively selected sites has been reported only on a few plant viruses (for
instance GLRaV-1 and GLRaV-4 strain 5) but absent positively selected sites
over the CP are more frequent. In fact, the comprehensive study of Chare and Holmes (2004) showed only three of 36 plant
virus species with low number of sites under positive selection into the CP
gene. High conservation levels of most CP (where up to 198 of 233 C-terminal
residues were inferred to be under negative selection) reflect a strong
purifying selection, probably maintaining some CP functions. For instance, a
putative nucleotide binding site was inferred in all the analyzed sequences in
the 175 to 182 positions, typically saturated of sites under strong negative
selection pressure. This selective behavior may be a consequence of structural
requirements.
In addition to structural
functions, CP of plant viruses is involved in vector specificity. The
virion-vector interaction of lettuce infectious yellow virus (LIYV, Crinivirus)
has been thoroughly studied, and the minor coat protein (CPm) determined
virions to vector binding (5). GLRaV-4, in opposite to
most Closteroviridae family members, lacks a CPm homologue, and the
viral particle appears to be completely covered by CP, as a homologous antibody
uniformly decorates the entire viral particle, whereas GLRaV-2 left an
undecorated tail (1). In consequence, the CP
replacing the absent CPm should constitute the vector binding determinant.
Generally, plant viruses are considered host generalists and vector-specific.
Nearly 60% of plant virus species are transmitted by a single vector, but less
than 10% of viral species infect one single host (24). GLRaV-1, -3 and -4 showed a particular behavior, given they
naturally infect only Vitis spp, whereas they are transmitted by up to
eleven different mealybugs and soft-scale insect species (9). Different isolates of GLRaV-4 have been reported as
transmissible with variable efficiency, or even not transmitted by six
pseudococids species belonging to two families. This biological behavior is
somewhat similar to the serological reactivity against Mabs previously reported
(11). In this work, we
confronted serological reactivity of local isolates of GLRaV-4 to a wide panel
of Mabs proved to be highly specific. Furthermore, the heterologous reaction
observed may be caused by multiple strain infection, rather than sensu
stricto heterologous serological reaction. Considering the variability and
antigenicity observed across the CP of GLRaV-4, we postulate that linear
epitopes reactive to Mab36-117, Mab 8-2, Mab3-1 and Mab3-3 are located in the
highly immunogenic N-terminal region of the protein. Previous research
demonstrated that for cucumber mosaic virus, a short epitope of five residues
exposed on the surface of the virion reacts with a Mab, essential for virus
transmissibility, as single residue mutations abolish both transmissibility and
reactivity against Mab (3, 19). However,
one single mutation can provide vector affinity advantages. If such mutation
increased transmission efficiency or augmented the number of vector species
having affinity for the virion, it would confer an impressive ecological
advantage compared with the wild-type population. This may explain the unusual
occurrence of positively selected sites in the CP of GLRaV-4. Conversely, the
highly conserved C-terminal region may be the result of structural conformation
of CP or strong CP interaction requirements in the virus replication cycle in
the plant. The SNAP2 analysis revealed that the region comprising the first 40
residues was mostly composed of amino acids whose substitution led to a neutral
function effect, whereas mutations in amino acids located around the putative
polynucleotide binding site (located in the C-terminal region) could have a
functional effect (Supplementary Figure 3). This
suggested biological significance of the observed sequence conservation.
The same evolutionary
behavior described (a variable N-terminal region with positively selected
sites) has been reported for bean yellow mosaic virus (Potyvirus). In that
species, Parella and Lanave demonstrated that one of the positively selected
sites identified belonged to a motif involved in CP-vector interaction, crucial
for transmissibility (23).
The significance behind
these observations in ampeloviruses can be assessed by an exhaustive study of
the transmission efficiency of different genetic variants in the presence of
different mealybug species. Recently, Rivadeneira et
al. (2022) reported differential incidence of GLRaV -3 and -4,
suggesting higher levels of GLRaV-3 linked to the occurrence of Planococcus
ficus. However, surprisingly in the presence of mealybugs, GLRaV-4
incidence remains quite low. In contrast, GLRaV-3 incidence is lower in Mendoza
Province (10, 17). Consequently,
assertive identification of viral strains should be considered for disease
impact, like in modelization approaches of vigour components in grapevine (14, 15).
Epitope prediction conducted
using BepiPred revealed high occurrence probability of a linear B-cell epitope
in the N-terminal region of CP, consistent with previous observations (6, 20). In addition to the implication in the
abovementioned transmissibility, some important immunological issues need
discussion. Considering that most available serological reagents for
characterizing GLRaV-4 isolates are monoclonal antibodies (11) with good reactivity against
denatured CP in Western blots, they might be directed against a linear epitope.
Moreover, given viral particles were applied in the native form during
immunization, these epitopes may be located on the virion surface. Considering
the most immunogenic region as a linear epitope (the most variable region),
monoclonal antibodies targeting these epitopes will not be useful for taxonomic
purposes at species level. This statement considers the identity level found in
the present study for GLRaV-4 strain 5 in the N-terminal region ranged from 57%
to 100% while for GLRaV-4 strain 6, it ranged from 47% to 100%. However, these
antibodies remain useful for strain discrimination.
Conversely, the antibodies
present in the commercially available reagent set for GLRaV-4 (2) appeared to target a conformational epitope (as they are
nonreactive against the denatured CP in Western blots) highly conserved and
probably located in the C-terminal region of CP. Since no available systems
predict conformational epitopes from the primary structure of the proteins,
this issue remains unresolved and warrants further research.
Conclusions
This work first reports a
linkage among the distinctive evolutionary behavior of the coat protein of
GLRaV-4 and biological properties of such protein, providing an alternative
point of view in the study of virus-vector interactions, transmissibility and ecology.
1.
Abou Ghanem-Sabanadzovic, N.; Sabanadzovic, S.; Gugerli, P.; Rowhani, A. 2012.
Genome organization, serology and phylogeny of Grapevine leafroll-associated
viruses 4 and 6: Taxonomic implications. Virus Research. 163(1): 120-128.
2.
Besse, S.; Bitterlin, W.; Gugerli, P. 2009. Development of an ELISA for
simutaneous detection of Grapevine leafroll associated virus -4, -5, -6, -7 and
-9. Extended abstracts 16th Meeting
of ICVG, Dijon, France. 296-298.
3.
Bowman, V. D.; Chase, E. S.; Franz, A. W.; Chipman, P. R.; Zhang, X.; Perry, K.
L.; Baker, T. S.; Smith, T. J. 2002. An antibody to the putative aphid
recognition site on cucumber mosaic virus recognizes pentons but not hexons.
Journal of Virology. 76(23): 12250-12258.
4.
Chare, E. R.; Holmes, E. C. 2004. Selection pressures in the capsid genes of
plant RNA viruses reflect mode of transmission. Journal of General Virology.
85(10): 3149-3157.
5.
Chen, A. Y.; Walker, G. P.; Carter, D.; Ng, J. C. 2011. A virus capsid
component mediates virion retention and transmission by its insect vector.
Proceedings of the National Academy of Sciences. 108(40): 16777-16782.
6.
Esteves, F.; Teixeira Santos, M.; Eiras-Dias, J. E.; Fonseca, F. 2012.
Occurrence of grapevine leafroll associated virus 5 in Portugal: genetic
variability and population structure in field-grown grapevines. Archives of
Virology. 157: 1747-1765.
7.
Esteves, F.; Santos, M. T.; Eiras-Dias, J. E.; Fonseca, F. 2013. Molecular data
mining to improve antibody-based detection of Grapevine leafroll-associated
virus 1 (GLRaV-1). Journal of Virological Methods. 194(1-2): 258-270.
8.
Flamarique, S. S.; Vilanova Perez, A.; Luque, A. V.; Rodríguez Pardina, P. E.;
del Valle Di Feo, L. 2022. Advances in the etiology of sweet potato (Ipomoea
batatas (L.) Lam) yellow curling disease in Argentina. Revista de la
Facultad de Ciencias Agrarias. Universidad Nacional de Cuyo. Mendoza.
Argentina. 54(2): 107-116.
9.
Fuchs, M.; Marsella-Herrick, P.; Loeb, G. M.; Martinson, T. E.; Hoch, H. C.
2009. Diversity of ampeloviruses in mealybug and soft scale vectors and in
grapevine hosts from leafroll affected vineyards. Phytopathology. 99(10):
1177-1184.
10.
Gomez Talquenca, S.; Alonso, R.; Luna, F.; Lanza Volpe, M.; Buscema, F. 2023.
Occurrence of Nine Grapevine Viruses in Commercial Vineyards of Mendoza,
Argentina. Viruses. 15(1): 177.
11.
Gugerli, P. 2009. 25 years of serological identification of grapevine
leafroll-associated viruses: antiserum and monoclonal antibodies to GLRaV-1 to
GLRaV-9. In Extended Abstract 16th Meeting
of ICVG, Dijon, France. p. 24-28.
12.
Gugerli, P.; Ramel, M. E. 1993. Grapevine leafroll associated virus II analyzed
by monoclonal antibodies. In Extended abstracts 11th
meeting of ICVG, Montreux, Switzerland. p. 23-24.
13.
Gugerli, P.; Brugger, J. J.; Ramel, M. E. 1997. Identification immuno-chimique
de 6e virus associé à la maladie de l’enroulement de la vigne et amélioration
des techniques de diagnostic pour la sélection sanitaire en viticulture. Revue
Suisse de Viticulture, Arboriculture et Horticulture. 29(3): 137-142.
14.
Hugalde, I. P.; Agüero, C. B.; Barrios-Masias, F. H.; Romero, N.; Nguyen, A.
V.; Riaz, S.; Piccoli, P.; McElrone, A. J.; Walker, M. A.; Vila, A. F. 2020.
Modeling vegetative vigour in grapevine: unraveling underlying mechanisms.
Heliyon. 6(12), e05708.
15. Hugalde, I.; Paolinelli, M.; Agüero, C. B.; Riaz, S.; Gómez
Talquenca, S.; Walker, M. A.; Vila, H. 2021. Prioritization of vigor
QTL-associated genes for future genome-directed Vitis breeding. Revista de la Facultad de
Ciencias Agrarias . Universidad Nacional de Cuyo.
Mendoza. Argentina. 53(2): 27-35.
16.
Kosakovsky Pond, S. L.; Frost, S. D. 2005. Not so different after all: a
comparison of methods for detecting amino acid sites under selection. Molecular
biology and evolution. 22(5): 1208-1222.
17.
Lanza Volpe, M.; Moyano, S.; Lijavetzky, D.; Talquenca, S. G. 2015. Partial
molecular and biological characterization of Grapevine leafroll-associated
virus 2 isolates from Argentina. Journal of Plant Pathology. 97(2): 349-355.
18.
Larsen, J. E. P.; Lund, O.; Nielsen, M. 2006. Improved method for predicting
linear B-cell epitopes. Immunome Research. 2(1): 1-7.
19.
Liu, S.; He, X.; Park, G.; Josefsson, C.; Perry, K. L. 2002. A conserved capsid
protein surface domain of Cucumber mosaic virus is essential for efficient
aphid vector transmission. Journal
of Virology . 76(19):
9756-9762.
20.
Maliogka, V. I.; Dovas, C. I.; Katis, N. I. 2008. Evolutionary relationships of
virus species belonging to a distinct lineage within the Ampelovirus genus. Virus Research . 135(1): 125-135.
21.
Martelli, G. P.; Ghanem-Sabanadzovic, N. A.; Agranovsky, A. A.; Rwahnih, M. A.;
Dolja, V. V.; Dovas, C. I.; Fuchs, M.; Gugerli, P.; Hu, J. S.; Jelkmann, W.;
Katis, N. I.; Maliogka, V. I.; Melzer, M. J.; Menzel, W.; Minafra, A.; Rott, M.
E.; Rowhani, A.; Sabanadzovic, S.; Saldarelli, P. 2012. Taxonomic revision of
the family Closteroviridae with special reference to the grapevine
leafroll-associated members of the genus Ampelovirus and the putative species
unassigned to the family. Journal of Plant Pathology. 7-19.
22.
Martin, D. P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. 2015. RDP4:
Detection and analysis of recombination patterns in virus genomes. Virus
Evolution. 1:(1).
23.
Parrella, G.; Lanave, C. 2009. Identification of a new pathotype of Bean yellow
mosaic virus (BYMV) infecting blue passion flower and some evolutionary
characteristics of BYMV. Archives of Virology. 154: 1689-1694.
24.
Power, A. G. 2008. Community ecology of plant viruses. Plant Virus Evolution.
15-26.
25.
Rivadeneira, M.; Galván, M. Z.; Abán, M.; Semke, R. E.; Rivadeneira, J.; Lanza
Volpe, M.; Gomez Talquenca, S. 2022. Survey for major grapevine viruses in
commercial vineyards of Northwestern Argentina. Plants. 11(13): 1720.
26.
Saldarelli, P.; Cornuet, P.; Vigne, E.; Talas, F.; Bronnenkant, I.; Dridi, A.
M.; Andret-Link, P.; Boscia, D.; Gugerli, P.; Fuchs, M.; Martelli, G. P. 2006.
Partial characterization of two divergent variants of grapevine
leafroll-associated virus 4. Journal of Plant Pathology. 203-214.
27.
Savino, V. 1993. ‘Extraction of closterovirus from grapevine tissues’, in G. P.
Martell (ed.), Graft-transmissible diseases of grapevines. Handbook for
detection and diagnosis (Rome: FAO). 239.
28.
Swofford, D. L. 2002. PAUP: phylogenetic analysis using parsimony (and other
methods), version 4.0 beta. http://paup.csit.fsu.edu/
29.
Thompson, J. R.; Fuchs, M.; Perry, K. L. 2012. Genomic analysis of Grapevine
leafroll associated virus-5 and related viruses. Virus Research .
163(1): 19-27.
30.
Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.;
Hönigschmid, P.; Schafferhans, A.; Roos, M.; Bernhofer, M.; Richter, L.;
Ashkenazy, H.; Punta, M.; Schlessinger, A.; Bromberg, Y.; Schneider, R.;
Vriend, G.; Sander, C.; Ben-Tal, N.; Rost, B. 2014. PredictProtein-an open
resource for online prediction of protein structural and functional features.
Nucleic Acids Research, 42(W1): W337-W343.
31. Zwickl, D. J. 2006. Genetic algorithm approaches for the
phylogenetic analysis of large biological sequence datasets under the maximum
likelihood criterion. The University of Texas at Austin. 1-125.
Supplementary
tables and figures